Structural Genomics Consortium
What are the CACHE challenges?
The inaugural CACHE challenge was launched in December 2021. Organized by the Structural Genomics Consortium (SGC), CACHE (Critical Assessment of Computational Hit-finding Experiments) is a public–private partnership benchmarking initiative to enable the development of computational methods “to compare and improve small-molecule hit-finding algorithms through cycles of prediction and experimental testing.”
The hope is that as participants test different computational methods within the CACHE challenges, they’ll discover novel small molecules targeting proteins related to human disease (i.e., new starting points for drug discovery). The goal is to launch three different “challenges” every year, each focused on a different protein target. (You can read more about the CACHE roadmap in Nature Reviews Chemistry.)
Participation is not guaranteed. Groups, including ours, submit applications which are peer-reviewed. As of the third CACHE challenge, the Canadian government has graciously supported the involvement of Canadian SMEs, facilitated most recently by Conscience. We are thankful for the financial contributions of these institutions.
Why we’re participating
As a Research-as-a-Service (RaaS) start-up in computer-aided drug design, MFI has a clear focus: to help kickstart drug discovery programs around the world with our software and expertise. It’s social impact that drives our mission (read more about that here) and we are in business to help people, to further science, and to make drug discovery more efficient. However, because much of MFI’s work and contributions are bound by NDA, we cannot share the information publicly. This is why the CACHE framework is energizing and exciting to our team: we have the opportunity to collaborate beyond typical confidentiality limitations and to support researchers by sharing what we do and how we do it in a broader context. Furthermore, we aim to push the boundaries of “conventional” methods and develop new approaches and workflows as part of each challenge.
It’s all about collaboration, not competition
To us, the value of CACHE is not about finding the most efficient computational methods for hit-finding as much as guiding technological improvements in future computational approaches to drug discovery. After all, every organization that is participating in CACHE has tools and expertise to benefit any given drug discovery program. That is why we’re approaching our participation in CACHE as collaborative rather than competitive, embracing the program as an opportunity to push ourselves and each other.
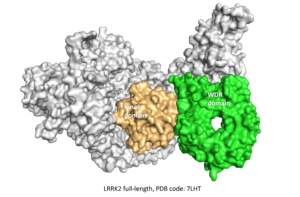 ⇒ WDR domain of LRRK2
⇒ WDR domain of LRRK2
⇒ Parkinson’s Disease
⇒ Consensus scoring method
In March 2022, Molecular Forecaster was selected with 22 other teams to participate in the first CACHE challenge supported by the Michael J. Fox Foundation. This inaugural challenge takes aim at LRRK2, the most commonly mutated gene in familial Parkinson’s Disease by targeting the WD40 repeat (WDR) domain of LRRK2.
To find hits for this challenge we first rebuilt the missing parts of the crystal structure targeted (PDB ID: 6DLO, green) and optimized our reconstructed model using molecular dynamic simulations to extract the most common conformation of the LRRK2 domain. We then filtered two libraries from the Enamine and Mcule vendors according to CACHE white paper guidelines to prepare the sub-library of compounds for virtual screening. After completing the virtual screening campaign using our in-house docking program FITTED, we used four separate ranking approaches based on docking scores, ML scores, quantum mechanics scoring function, and visualization to select the 100 compounds we submitted for further testing to the CACHE committee.
Our manuscript can be found here.
 ⇒ RNA binding site of SARS-Cov-NSP13
⇒ RNA binding site of SARS-Cov-NSP13
⇒ Coronaviruses
⇒ Consensus-scoring method, and templated docking
The second CACHE Challenge was launched in June 2022 and focused on finding ligands targeting the RNA binding site of the underexplored NSP13 helicase, which is the most conserved site in SARS-CoV-2 and across coronaviruses. For this challenge, 24 teams were selected to participate and submit up to 100 compounds.
The first step was to review and analyze the multiple structures of the helicase available in the PDB, enabling us to determine two possible binding sites from fragment-bound structures. We then developed different filtering protocols of an Enamine library to prepare sub-libraries of ligands suitable for each binding site. In addition, we prepared a third sub-library comprised of compounds matching the co-crystallized fragments to perform templated docking. The three sub-libraries were then docked to the NPS13 structures, and 100 compounds were chosen based on a consensus decision using docking scores, ML-based scores, quantum mechanics-based scoring function, and compound visualization.
The manuscript capturing our work can be found here.
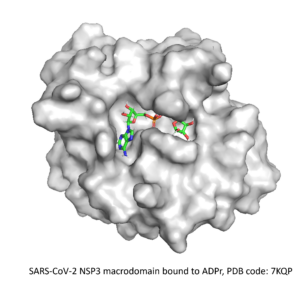 ⇒ Macrodomain of SARS-CoV-2 NSP3
⇒ Macrodomain of SARS-CoV-2 NSP3
⇒ Severe acute respiratory syndrome coronavirus
⇒ AI and knowledge-based method
In November 2022, SGC launched the third CACHE challenge, which gathered 25 teams to predict compounds for the macrodomain of SARS-CoV-2 NSP3. With hundreds of fragment- and inhibitor-bound structures already reported, participants were asked to predict compounds with novel chemical templates that can compete with the endogenous substrate, ADP-ribose.
After understanding the structural characteristics of the protein binding site from the available structures in the PDB, we selected many interesting fragments as starting points for our dual strategy. We designed potential binders for our knowledge-based fragment merging approach and implemented a recurrent neural network for our AI-guided fragment expansion one. The molecule templates generated by both methods were then used to search for analogs in the Enamine REAL database, which were then virtually screened against the target protein. Finally, 100 compounds were proposed for purchase and testing.
Our manuscript can be found here.
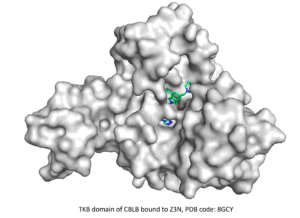
⇒ TKB domain of CBLB
⇒ Cancers
⇒ Pose-refinement with MD method
The fourth CACHE challenge was launched in March 2023 and focused on finding ligands targeting the TKB domain of the E3 ubiquitin-protein ligase CBLB. This target being well-known, hundreds of compounds were reported in the patent literature, one of which gives key information about the conformational structure upon binding. The 23 teams of participants were asked to predict molecules that bind to the same pocket and compete with the co-crystallized compound while representing novel chemical templates and respecting a given disassociation constant.
The set of patented inhibitors provided by CACHE was used for a retrospective benchmarking that served both to sanity-check our methodology and to develop multiple pharmacophoric models. After curating the library of compounds to be tested based on a SAR analysis of the patented inhibitors, we filtered it to ensure chemical and structural diversity and to meet common medicinal guidelines. We virtual screened the ligand library against our pharmacophore models to further reduce the number of ligands to screen against the target protein. The ligand binding modes of the latter screen were analyzed, and 200 diverse molecules were selected for molecular dynamics simulations and MM/PBSA analysis, which ultimately guided the selection of the 100 submitted molecules.
Our manuscript can be found here.
⇒ Melanine-concentrating hormone receptor
⇒ Obesity, Depression
⇒ Consensus scoring, and active learning
The fifth CACHE challenge was launched in December 2023 and focused on finding novel antagonists of the Melanin-Concentrating Hormone Receptor (MCHR1). The CACHE committee provided the participants with a chemical diverse array of reported compounds (patented or not), however no experimental structure was available for this target. The 24 teams selected for this challenge had to predict up to a 100 chemically novel antagonists for MCHR1 below 30 µM.
The set of representative MCHR1 antagonists provided by CACHE was docked to the protein model we built based on the predicted AlphaFold structure. We ran molecular dynamics simulations on two post-docking complexes and extracted representative protein structures. We further used these structures for retrospective benchmarking as well as for the development of pharmacophore models. Simultaneously we developed a suitable library for prospective screening, using the Enamine REAL database. We applied active learning (AL) to identify suitable molecules for docking, and screened against the pharmacophore models. We finally docked the molecule shortlisted by the dual AL and pharmacophore screenings, numerically and visually analyzed them, and selected 100 molecules for testing.
Stay tuned for our manuscript!
⇒ Triple Tudor Domain of SET Domain Bifurcated 1 
⇒ Epigenetic regulation
⇒ Consensus scoring method and high throughput molecular dynamics
In May 2024, SGC launched the sixth CACHE challenge focused on the histone groove of the SETDB1 triple Tudor domain (TTD). Recent crystallographic studies have revealed that ligands can engage one or more of the three TTD subcavities. The participants were therefore asked to predict chemically novel ligands occupying one or multiple subcavities of the histone binding groove of the target.
We first collected a set of SETDB1 structures bound to peptides, small molecules and fragments and performed self- and cross- docking using our internal program FITTED to respectively assess its effectiveness and select structures for further analysis. Five structures were then used to build structure-based pharmacophore models, and we screened the Enamine REAL library database against each model to pick compounds that aligned best with those pharmacophores. Subsequently, we performed docking-based virtual screenings. We filtered and clustered the results based on specific interactions and sent this dataset to HtuO, our partner on this project, for high-throughput molecular dynamics using their proprietary technology. Together, we selected 100 compounds for purchase.
Stay tuned for our manuscript!