Structural Genomics Consortium
Quels sont les défis CACHE ?
Le premier défi défi CACHE a été lancé en décembre 2021. Organisé par le Consortium de génomique structurelle (SGC)CACHE (Critical Assessment of Computational Hit-finding Experiments) est une initiative d’évaluation comparative en partenariat public-privé visant à permettre le développement de méthodes informatiques « pour comparer et améliorer les algorithmes de recherche de petites molécules à travers des cycles de prédiction et de tests expérimentaux ».
L’espoir est qu’en testant différentes méthodes de calcul dans le cadre des défis CACHE, les participants découvriront de nouvelles petites molécules ciblant des protéines liées à des maladies humaines (c’est-à-dire de nouveaux points de départ pour la découverte de médicaments). L’objectif est de lancer trois « défis » différents chaque année, chacun étant axé sur une cible protéique différente. (Vous pouvez en savoir plus sur la feuille de route de CACHE dans Nature Reviews Chemistry.)
La participation n’est pas garantie. Les groupes, dont le nôtre, soumettent des candidatures qui sont évaluées par des pairs. Depuis le troisième défi CACHE, le gouvernement canadien a gracieusement soutenu la participation des PME canadiennes, facilitée tout récemment par Conscience. Nous remercions ces institutions pour leur contribution financière.
Pourquoi nous participons
En tant que start-up de recherche sous forme de service (RaaS) dans le domaine de la conception de médicaments assistée par ordinateur, MFI a un objectif clair : aider à lancer des programmes de découverte de médicaments dans le monde entier grâce à nos logiciels et à notre expertise. C’est l’impact social qui est le moteur de notre mission (pour en savoir plus, cliquez ici). ici) et nous sommes en affaires pour aider les gens, faire avancer la science et rendre la découverte de médicaments plus efficace. Cependant, comme une grande partie du travail et des contributions d’MFI est soumise à un accord de confidentialité, nous ne pouvons pas partager l’information publiquement. C’est pourquoi le cadre créé par CACHE est à la fois stimulant et passionnant pour notre équipe : nous avons la possibilité de collaborer au-delà des limites de confidentialité habituelles et de soutenir les chercheurs en partageant ce que nous faisons et comment nous le faisons dans un contexte plus large. De plus, nous visons à repousser les limites des méthodes « conventionnelles » et à développer de nouvelles approches et de nouvelles procédures de travail dans le cadre de chaque défi.
Il s’agit de collaboration et non de compétition
Pour nous, l’intérêt de CACHE n’est pas uniquement de trouver les méthodes informatiques les plus efficaces pour la recherche de hits, mais aussi de guider les améliorations technologiques des approches computationnelles pour la découverte de médicaments. Après tout, chaque organisation participant à CACHE dispose d’outils et d’une expertise qui peuvent profiter à n’importe quel programme de découverte de médicaments. C’est pourquoi nous abordons notre participation à CACHE sous l’angle de la collaboration plutôt que de la concurrence, en considérant le programme comme une occasion de nous dépasser et de nous dépasser les uns les autres.
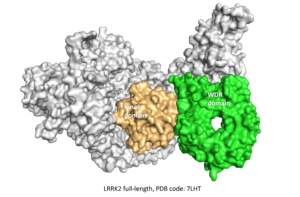 ⇒ Domaine WDR de LRRK2
⇒ Domaine WDR de LRRK2
⇒ Maladie de Parkinson
⇒ Méthode d’évaluation par consensus
En mars 2022, Molecular Forecaster a été sélectionné avec 22 autres équipes pour participer au premier défi CACHE soutenu par la Fondation Michael J. Fox. Ce défi inaugural vise LRRK2, le gène le plus souvent muté dans la maladie de Parkinson, en ciblant le domaine répétitif WD40 (WDR) de LRRK2.
Pour trouver des réponses à ce défi, nous avons d’abord reconstruit les parties manquantes de la structure cristalline ciblée (PDB ID : 6DLO, vert) et optimisé notre modèle reconstruit en utilisant des simulations de dynamique moléculaire pour extraire la conformation la plus courante du domaine LRRK2. Nous avons ensuite filtré deux bibliothèques des fournisseurs Enamine et Mcule selon les directives du manuel de CACHE pour préparer la sous-bibliothèque de composés pour le criblage virtuel. Après avoir terminé la campagne de criblage virtuel à l’aide de notre programme de docking interne FITTED, nous avons utilisé quatre approches de classement distinctes basées sur les scores de docking, les scores d’apprentissage machine, la fonction de score relative à la mécanique quantique et la visualisation pour sélectionner les 100 composés que nous avons soumis au comité de CACHE en vue d’un examen plus approfondi.
Notre manuscrit peut être consulté ici.
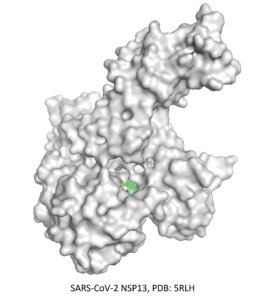 ⇒ Site de liaison de l’ARN du SARS-Cov-2 NSP13
⇒ Site de liaison de l’ARN du SARS-Cov-2 NSP13
⇒ Coronavirus
⇒ Méthode d’évaluation par consensus et docking basé sur un modèle
Le deuxième défi CACHE a été lancé en juin 2022 et s’est concentré sur la recherche de ligands ciblant le site de liaison à l’ARN de l’hélicase NSP13, un site peu exploré, qui est le site le plus conservé dans le SARS-CoV-2 et dans l’ensemble des coronavirus. Pour ce défi, 24 équipes ont été sélectionnées pour participer et soumettre jusqu’à 100 composés.
La première étape a consisté à examiner et à analyser les multiples structures de l’hélicase disponibles dans la PDB, ce qui nous a permis de déterminer deux sites de liaison possibles à partir des structures liées à des fragments. Nous avons ensuite mis au point différents protocoles pour filtrer une bibliothèque d’Enamine afin de préparer des sous-bibliothèques de ligands adaptés à chaque site de liaison. En outre, nous avons préparé une troisième sous-bibliothèque composée de molécules correspondant aux fragments cocristallisés afin d’effectuer un docking basé sur un modèle. Les trois sous-bibliothèques ont ensuite été dockées aux structures NPS13 et 100 composés ont été sélectionnés sur la base d’une décision consensuelle utilisant les scores de docking, les scores basés sur l’apprentissage automatique, la fonction de notation basée sur la mécanique quantique et la visualisation des composés.
Le manuscrit présentant notre travail peut être consulté ici.
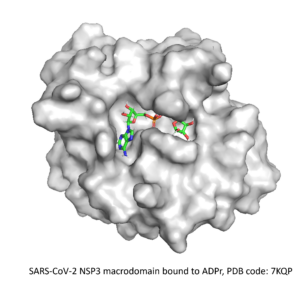 ⇒ Macrodomaine du SARS-CoV-2 NSP3
⇒ Macrodomaine du SARS-CoV-2 NSP3
⇒ Coronavirus du syndrome respiratoire sévère aigu
⇒ Méthode basée sur l’IA et la connaissance
En novembre 2022, le SGC a lancé le troisième défi CACHEqui a rassemblé 25 équipes chargées de prédire des composés pour le macrodomaine du SARS-CoV-2 NSP3. Des centaines de structures liées à des fragments et à des inhibiteurs ayant déjà été rapportées, il a été demandé aux participants de prédire des composés avec de nouveaux modèles chimiques pouvant entrer en compétition avec le substrat endogène, l’ADP-ribose.
Après avoir compris les caractéristiques structurelles du site de liaison de la protéine à partir des structures disponibles dans la PDB, nous avons sélectionné de nombreux fragments intéressants comme points de départ de notre double stratégie. Nous avons conçu des liants potentiels pour notre approche de fusion de fragments basée sur la connaissance et mis en œuvre un réseau neuronal récurrent pour notre approche d’expansion de fragments guidée par l’IA. Les modèles de molécules générés par les deux méthodes ont ensuite été utilisés pour rechercher des analogues dans la base de données Enamine REAL, qui ont ensuite été virtuellement criblés contre la protéine cible. Enfin, 100 composés ont été proposés pour être achetés et testés.
Notre manuscrit peut être consulté ici.
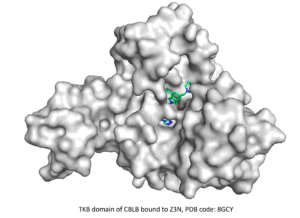
⇒ Domaine TKB de CBLB
⇒ Cancers
⇒ Raffinement de poses par dynamique moléculaire
Le quatrième défi CACHE a été lancé en mars 2023 et s’est concentré sur la recherche de ligands ciblant le domaine TKB de la E3 ubiquitine-protéine ligase CBLB. Cette cible étant bien connue, des centaines de composés ont été rapportés dans la littérature brevetée, dont l’un fournit des informations essentielles sur la structure conformationnelle lors de la liaison. Les 23 équipes de participants ont été invitées à prédire des molécules qui se lient à la même poche et entrent en compétition avec le composé co-cristallisé tout en représentant de nouveaux modèles chimiques et en respectant une constante de dissociation donnée.
L’ensemble des inhibiteurs brevetés fournis par CACHE a été utilisé pour un benchmarking rétrospectif qui a servi à la fois à vérifier le bien-fondé de notre méthodologie et à développer de multiples modèles pharmacophoriques. Après avoir constitué la bibliothèque de composés à tester sur la base d’une analyse SAR des inhibiteurs brevetés, nous l’avons filtrée afin de garantir la diversité chimique et structurelle et de respecter les directives médicinales courantes. Nous avons procédé à un criblage virtuel de la bibliothèque de ligands par rapport à nos modèles de pharmacophore afin de réduire encore le nombre de ligands à cribler par rapport à la protéine cible. Les modes de liaison des ligands de ce dernier écran ont été analysés et 200 molécules diverses ont été sélectionnées pour les simulations de dynamique moléculaire et l’analyse MM/PBSA, qui ont finalement guidé la sélection des 100 molécules soumises.
Notre manuscrit peut être consulté ici.
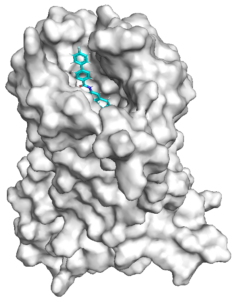
⇒ Récepteur de l’hormone de concentration de mélanine
⇒ Obésité, dépression
⇒ Notation consensuelle et apprentissage actif
Le cinquième défi CACHE a été lancé en décembre 2023 et s’est concentré sur la recherche de nouveaux antagonistes du récepteur de l’hormone de concentration de la mélanine (MCHR1). Le comité CACHE a fourni aux participants un ensemble de composés chimiques diversifiés (brevetés ou non), mais aucune structure expérimentale n’était disponible pour cette cible. Les 24 équipes sélectionnées pour ce défi devaient prédire jusqu’à 100 antagonistes chimiquement nouveaux pour MCHR1 en dessous de 30 µM.
L’ensemble des antagonistes représentatifs de MCHR1 fournis par CACHE a été docké au modèle de protéine que nous avons construit à partir de la structure AlphaFold prédite. Nous avons effectué des simulations de dynamique moléculaire sur deux complexes après docking et extrait des structures protéiques représentatives. Nous avons ensuite utilisé ces structures pour effectuer un benchmarking rétrospectif ainsi que pour le développement de modèles pharmacophores. Simultanément, nous avons développé une bibliothèque appropriée pour le criblage futur, en utilisant la base de données Enamine REAL . Nous avons appliqué L’apprentissage actif (AL) a permis d’identifier les molécules appropriées pour le docking, et a été comparé aux modèles de pharmacophores. Nous avons finalement docké les molécules présélectionnées par les criblages basés sur l’apprentissage actif et les pharmacophores, nous les avons analysées numériquement et visuellement, et nous avons sélectionné 100 molécules pour les tester.
Restez à l’affût de notre manuscrit !
⇒ Domaine triple Tudor de SET Domaine bifurqué 1 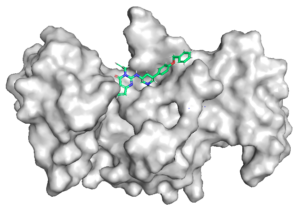
⇒ Régulation épigénétique
⇒ Notation consensuelle et dynamique moléculaire à haut débit
En mai 2024, le SGC a lancé le sixième défi CACHE consacré au sillon histonique du triple domaine de Tudor (TTD) de SETDB1. Des études cristallographiques récentes ont révélé que des ligands peuvent se fixer dans une ou plusieurs des trois sous-cavités du TTD. Les participants ont donc été invités à prédire des ligands chimiquement innovants occupant une ou plusieurs sous-cavités du sillon de liaison de l’histone de la cible.
Nous avons d’abord recueilli un ensemble de structures SETDB1 liées à des peptides, des petites molécules et des fragments, et nous avons effectué du self- et cross- docking à l’aide de notre programme interne FITTED afin d’évaluer son efficacité et de sélectionner des structures pour les prochaines analyses. Cinq structures ont ensuite été utilisées pour construire des modèles pharmacophores basés sur la structure et nous avons criblé la base de données de la bibliothèque Enamine REAL sur chaque modèle pour sélectionner les composés qui s’alignaient le mieux avec ces pharmacophores. Ensuite, nous avons effectué des criblages virtuels basés sur le docking. Nous avons filtré et regroupé les résultats sur la base d’interactions spécifiques et envoyé cet ensemble de données à HtuO, notre partenaire sur ce projet, pour la dynamique moléculaire à haut débit en utilisant leur technologie propriétaire. Ensemble, nous avons sélectionné 100 composés à acheter.
Restez à l’affût de notre manuscrit !